|
|
MOLECULAR VECTOR MACHINE OF PROTEINS Personal page number 3 |
|
Book V. Karasev and V. Luchinin "Introduction to the design of bionic
nanosystems"
Sites V.A. Karasev: Principles of topological
New Spatial structure of the canonical set of amino acids http://amino-acids-20.narod.ru Aid to the atheist: A new theory explaining essence and origin of
life |
Dear visitors! I thank, that you have glanced on my
page People used to say: all roads lead to Rome. Research
conducted by scientists at the first sight, entirely independent direction,
also sometimes lead to the same point of intersection. The page, proposed to Your attention, is the third
of a series of WEB pages devoted to the problem of topological coding of
proteins, planned by the author. The first page http://genetic-code.narod.ru opens
this series and has the common name "Principles of topological coding of
proteins." It focuses primarily on the structure of the genetic code and
the problem of assignment a triplet - an amino acid. The results obtained in
Section 4.4. (http://genetic-code.narod.ru/symm_conform.htm
) directly deduce to the idea of molecular vector
machine. On the second page http://amino-acids-20.narod.ru, which is called "The spatial
structure of the canonical set of amino acids",
the spatial structure of the canonical set of amino acids is examined, being
presented in the form of a dodecahedron. Trying to make sense of why there is
such a structure, not the other, has also led us to the idea of molecular
vector machine. Thus, two independent paths of investigation have
led us to the same idea of the molecular vector machine, which this page is devoted to. The results of this work a few ahead of several colleagues,
owing to what we practically have nobody to refer, except as to own results [1-5], and attempts to continue these studies in the original direction of
the two independent masters [6-8]. However, this
does not exclude that in the near future the proposed path of research
interest to colleagues of different specialties, primarily physicists,
mathematicians and molecular biologists, and these studies will be continued. |

|
1. Introduction |
|
|
|
|
|
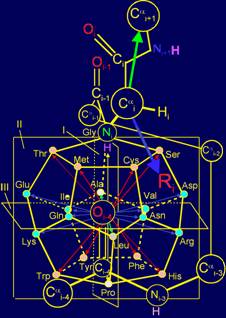
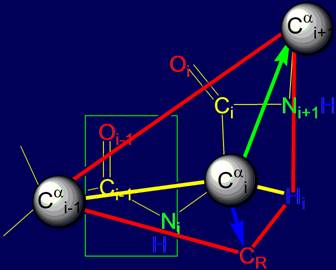
The
object of analysis is the area of bond NiH….Oi-4=C in the protein pentafragment |
|
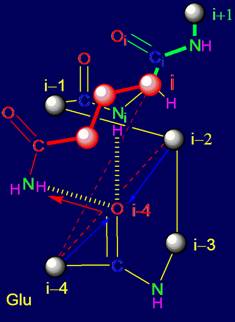
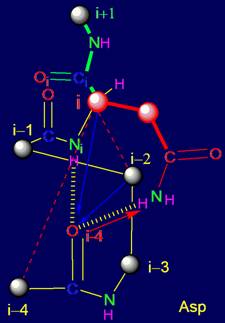
In “Introduction” the subject of the
forthcoming analysis is defined - area of bond
NiH….Oi-4=C in protein pentafragment and defined the
stages of its consideration
2. Stages of construction of the molecular vector machine of
proteins
|
|
|
|
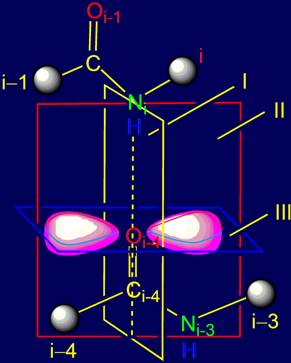
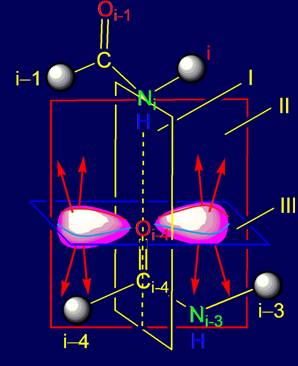
Results:
1.
In the area of bond NiH….Oi-4=C from a position of quantum chemistry
three planes of symmetry dividing sp2--hybridized clouds on symmetric parts are
introduced.
2.
Taking into account the allocated planes
of symmetry four groups of vectors of action are received.
3.
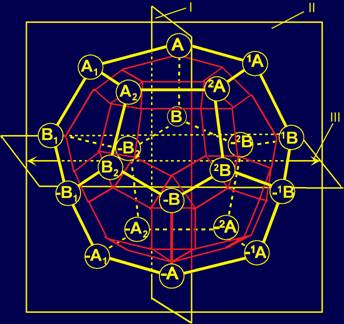
The model of the molecular vector machine
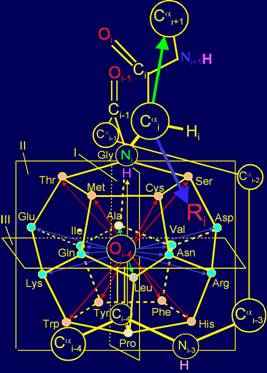
(МVМ) is offered, consisting:
- From system of the vectors localized in structure of a dodecahedron;
- The canonical set of side chains of amino acids (exchangeable physical
operators);
- i-th tetrahedral alpha-carbon atom to which exchangeable side chains
of amino acids are attached;
- Amino acid pentafragment of protein main chain.
3.Properties of
parts of the molecular vector machine of proteins
|
|
|
|
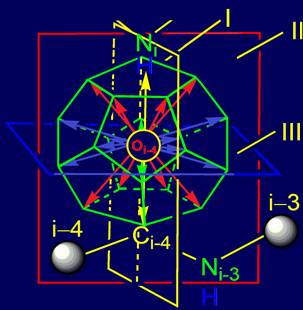
Results:
1.
The analysis of the vectors from the
position of group-theoretic approach is carried out. It is concluded that 20 of
the vectors inside the dodecahedron form a mathematical group containing
neutral and inverse elements.
2.
On the basis of group-theoretical
approach the canonical set of side chains of amino acids are considered as a
group of irreducible representations of the vectors.
3.
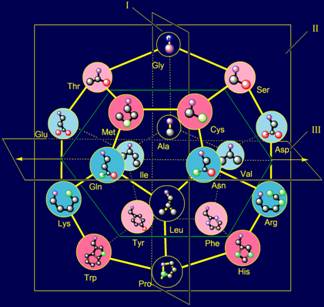
From the standpoint of of representations
of MVM and the group-theoretic approach, an explanation of the principles used
for construction of spatial structure of the canonical set of amino acids on
the dodecahedron (http://amino-acids-20.narod.ru/AA_dodecahedron.htm)
is offered:
- The principle of an arrangement of side chains from top to bottom in
order of increasing size;
- The principles of the side chains of amino acids antisymmetry.
It is concluded that these principles are related with a reconstruction
them of symmetric vectors of action in МVМ.
4.
The analysis of the properties of the
i-th tetrahedral alpha-carbon atom, a kind of "yoke", which specifies
the direction of growth of the polypeptide chain, depending on the type of side
chain with respect to this atom is carried out.
5.
It is assumed that the analysis of this
region may be promising for the development of methods for predicting the
secondary structure of proteins.
4. Two-layer
model of the molecular vector machine
|
4.2. Romboikosododekahedron and dodecahedron as a elements of
MVM |
||
|
|
|
|
|
|
|
|
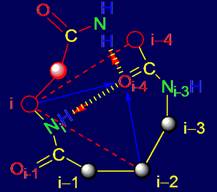
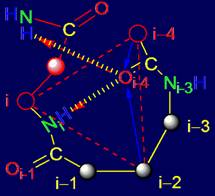

Results:
1.
In
order to resolve the contradiction between the number of side chains (20) and the
number of coding triplets (61) a two-layer model of the molecular vector
machine is proposed.
2.
It
is supposed that in addition to the dodecahedron, as a polyhedron, occupying
the lower level of MBM can serve romboikosododekahedron.
5. The conclusion. Practical application of concepts of molecular vector
machine
In concepts of the molecular vector machine it is possible to allocate
two aspects. On the one hand, they are, as it often happens in a science, a
formal abstraction ((similar to a Carnot heat engine), convenient for the
description and an explanation of some the biological facts (for example, the
nature of of the canonical set of amino acids).
On the other hand, these concepts are related to co-translational
mechanism for the formation of the secondary structure of proteins [5], implying that the formation of the secondary
structure of proteins occurs in the ribosome at the moment of its synthesis.
For this reason they are quite useful as a specific machine, carrying out this
process in the ribosome. Of course, this mechanism is still hypothetical, but
it can be a guide for experts, both theorists and experimentalists, and serve
as a starting point for further research in different directions.
In addition to the elements analyzed in Section 3, the structure of MVM also includes fragments of the five amino acids
(pentafragments). In real protein amino acid sequence and structure of
pentafragments bonds is the fixed reflexion of results of work МVМ.
For this reason, besides the theoretical analysis of properties of the elements
of MVM, it is possible to analyze pentafragments of proteins studied by X-ray
diffraction analysis, preserving their hydrogen bonds in the protein,
particularly in the secondary structure. We carried out such an analysis.
Its details, as we believe, can represent a wide interest. In the future, at the end of the cycle of works,
we expect to create a special page for the study and exposition of this
approach.
DURING THE PERFORMED ANALYSIS FOLLOWING
RESULTS HAVE BEEN RECEIVED::
-
16 classes of pentafragments, 8 of which are major and 8 – minor are found.
-
The spatial structure for each class, isomorphic to the Boolean hypercube B6
is proposed.
-
A complete structure of all pentafragments described by Boolean hypercube B4
is constructed, in which vertices hypercubes B6 are located.
For
those who wish to familiarise with the received results, we inform, that they
are published in works in [9-11],
are available for reading and downloading.
On
the basis of these results DATABASE of PENTAFRAGMENTS [12] has been created, and a method of predicting the secondary structure
of proteins [13], which is also
available for review and download, is
developed. This method can be performed both manually and by computer program
PREDICTOR [14]. All these
materials are integrated and can serve as a subject of licence sale. With
questions on acquisition of the licence interested persons can address to the
author via this page.
In
proteins, which petafragments have been used for database creation, accuracy of
a prediction of secondary structure makes 99%.
Now
the demand on a methods of designing of primary structure of the protein with a
given secondary structure is submitted. While it is at an examination stage.
We wish you the further scientific impressions!
Address
for connection: vector-machine@narod.ru
ã The Work
was carried out at the Centre of Microtechnologies and Diagnostics (CMID) of
the St.-Petersburg State Electrotechnical University "LETI", at
financial support of University
Acknowledgements:
I express my sincere gratitude to A.I. Belyaev for help
in editing the English version of this page.